Le Réseau GénoPsy : Troubles du comportement d’origine génétique, est un réseau national composé de 5 Centres de Référence Maladies Rares (CRMR) et de 6 Centres de Compétence Maladies Rares (CCMR). Le CRMR GénoPsy Lyon situé à l’hôpital du Vinatier en est le centre coordonnateur.
Labellisé en 2023 dans le cadre du nouveau Plan National maladies rares (PNMR3), il est porté par la filière de santé AnDDi-Rares.
Le réseau GénoPsy a pour objectif principal d’initier et de faciliter la collaboration entre les différents acteurs régionaux, nationaux et internationaux impliqués dans la recherche, le diagnostic, la formation et le soutien des personnes atteintes de maladies rares d’origine génétique avec des manifestations psychiatriques ainsi que de leur famille.
Description du poste
Placé·e sous la direction de la coordinatrice du réseau, la Professeure Caroline Demily, et en collaboration avec la cheffe de projet partenariats, le/la chef·fe de projet a pour mission de déployer la communication et d’animer le réseau.
Missions
Communication du Réseau GénoPsy
- Conception graphique
- Réalisation de vidéos
- Animation des réseaux sociaux
- Envoi d’une newsletter
- Création d’un site internet et de divers supports de communication
- Aide à la diffusion et à la reconnaissance de l’innovation clinique et scientifique
Animation du Réseau GénoPsy
- Organisation d’évènements scientifiques et grand public
- Faire le lien avec tous les CRMR et CCMR du réseau
- Mise en relation des acteurs du réseau pour construire des projets
- Aide à l’élaboration des projets de recherche
- Concourir à la bonne implantation des activités du réseau à l’échelon national
- Développer des synergies avec le Centre d’Excellence iMIND
- Veille scientifique
Formation
- Aide à l’élaboration et la promotion des formations du réseau
Gestion administrative
- Suivi de la base de données BAMARA
- Recueil des variables quantitatives de l’activité du CRMR Lyon
- Réalisation du bilan d’activité annuel, gestion des COPIL et rédaction des comptes-rendus
- Gestion des données / informations / indices qualité (recueil, saisie, analyse, diffusion, classement, suivi)
Profil recherché
- Avoir le sens de l’organisation et des priorités, pouvoir développer une vision intégrative
- Être dynamique et responsable de sa pratique
- Être disponible et flexible en termes d’horaires
- Être capable d’autonomie et de prise d’initiatives tout en respectant les règles de fonctionnement institutionnel
- Accompagner les porteurs de projet dans les démarches de recherche clinique (CCP, etc.)
- Pouvoir accompagner les autres professionnels dans les partenariats à tisser
- Capacités d’intégration au sein d’une équipe pluriprofessionnelle.
Formation souhaitée
- Bac + 5 – Équivalent chefferie de projet ou scientifique
Expérience professionnelle
- Expérience dans le champ de la gestion de projet, valorisation, communication. Une connaissance de la psychiatrie, des maladies rares et/ou des troubles du neurodéveloppement serait appréciée
Connaissances spécifiques attendues
- Maîtrise de Microsoft Office, la suite Adobe, CANVA et WordPress
- Aisance rédactionnelle ainsi qu’à l’oral
- Pouvoir se déplacer en dehors de Lyon (Permis B)
Informations sur le poste
- Type de contrat : CDD de 6 mois renouvelable avec possibilité de pérennisation
- Cycle hebdomadaire du lundi au vendredi
- Temps de travail : 100%
- Amplitudes horaires : 8h30 – 19h00
(en fonction des activités, disponibilité en dehors de ces créneaux possible) - Télétravail possible
- Lieu d’exercice : pôle HU-ADIS sur le site du Campus Hospitalier le Vinatier
- Salaire : selon profil et expérience
- Prise de poste : souhaitée début octobre 2024
Vous souhaitez candidater
- Pour postuler, envoyez un CV et une lettre de motivation avant le 31 août 2024 aux adresses suivantes: caroline.demily@ch-le-vinatier.fr – jennifer.beneyton@ch-le-vinatier.fr
- Préciser dans l’objet du mail : « POSTE Chef de projet – Réseau GénoPsy »


Qu’est-ce que l’UDN?
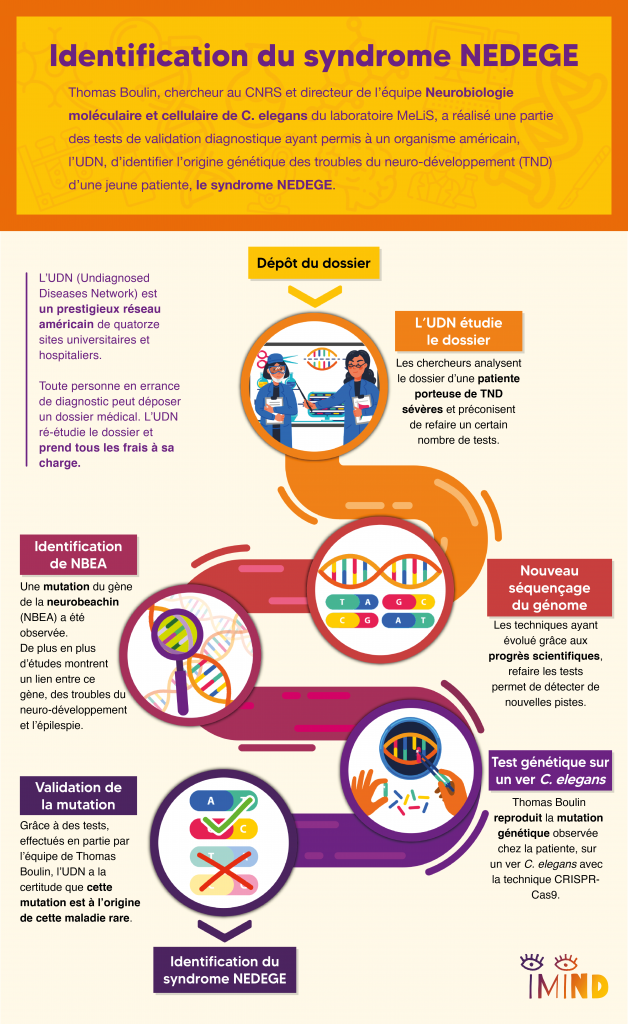
 L’UDN, pour Undiagnosed Diseases Network, est un réseau américain de quatorze sites universitaires et hospitaliers les plus prestigieux du pays, financé par le NIH. Un patient, ou sa famille, en errance diagnostique peut déposer auprès de cet organisme un dossier composé de tous ses antécédents et tests médicaux. Pour les patients pris en charge, l’UDN étudie tous les éléments du dossier et va actualiser certains tests ou faire des tests complémentaires, à la lumière des avancées technologiques et scientifiques les plus récentes. C’est entièrement pris en charge par le réseau, ce qui n’est pas négligeable lorsqu’on connaît le coût des frais médicaux aux États-Unis.
L’UDN, pour Undiagnosed Diseases Network, est un réseau américain de quatorze sites universitaires et hospitaliers les plus prestigieux du pays, financé par le NIH. Un patient, ou sa famille, en errance diagnostique peut déposer auprès de cet organisme un dossier composé de tous ses antécédents et tests médicaux. Pour les patients pris en charge, l’UDN étudie tous les éléments du dossier et va actualiser certains tests ou faire des tests complémentaires, à la lumière des avancées technologiques et scientifiques les plus récentes. C’est entièrement pris en charge par le réseau, ce qui n’est pas négligeable lorsqu’on connaît le coût des frais médicaux aux États-Unis.