IMINDgénétique
Sommaire
Identification d’un nouveau gène impliqué dans les troubles du neurodéveloppement : le gène DDX17
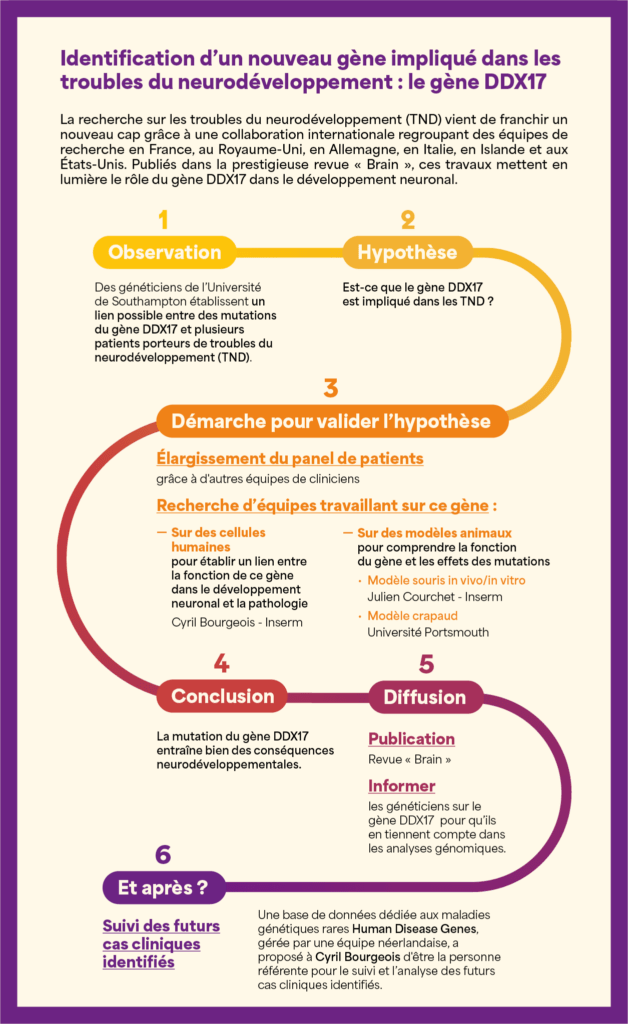
La recherche sur les troubles du neurodéveloppement (TND) vient de franchir un nouveau cap grâce à une collaboration internationale regroupant des équipes de recherche en France, au Royaume-Uni, en Allemagne, en Italie, en Islande et aux États-Unis. Publiés dans la prestigieuse revue Brain, ces travaux mettent en lumière le rôle du gène DDX17 dans le développement neuronal.
Retour sur cette découverte à la frontière entre génétique médicale et recherche fondamentale.

DDX17 : un gène au cœur de la régulation génique
Le gène DDX17 code pour une protéine appartenant à la famille des hélicases, qui sont impliquées dans la régulation de l’expression des gènes. Ces protéines jouent donc un rôle clé dans le contrôle des étapes menant à la synthèse des protéines, éléments constitutifs de la plupart de nos cellules humaines.
« Jusqu’à présent, nous étions surtout centrés sur de la recherche fondamentale autour des rôles de DDX17 dans la régulation des gènes », explique Cyril Bourgeois, qui a co-dirigé cette étude. « L’opportunité de collaborer avec des généticiens cliniques nous a permis d’explorer des applications directes de nos travaux. »
Les troubles du neurodéveloppement résultent d’une interaction complexe entre facteurs génétiques et environnementaux, dont les mécanismes précis restent encore largement méconnus. On estime aujourd’hui qu’environ 1 000 gènes sont impliqués dans ces troubles. Par ailleurs, l’influence de facteurs environnementaux est également étudiée pour mieux comprendre leur impact sur le développement cérébral. Grâce aux avancées en génomique, les chercheurs continuent d’affiner la liste des gènes impliqués et d’explorer les mécanismes biologiques sous-jacents, ouvrant ainsi la voie à de nouvelles approches diagnostiques et thérapeutiques. Cette étude sur le gène DDX17 permet de mettre en lumière un nouveau gène dont la mutation entraîne des conséquences neurodéveloppementales.
Une approche multidisciplinaire pour une découverte d’envergure
Tout a commencé quand une équipe de généticiens britanniques de l’Université de Southampton, dirigée par Sarah Ennis, a identifié le gène DDX17 comme un candidat d’intérêt dans les troubles du neurodéveloppement (TND). Grâce aux données issues du projet « 10 000 Genomes », ils ont établi un lien possible entre des mutations du gène DDX17 et plusieurs patients porteurs de TND, présentant un tableau clinique caractérisé notamment par une déficience intellectuelle, un retard de la parole et du langage, et un retard moteur, associé parfois à un syndrome autistique. Cependant, cette hypothèse nécessite une validation scientifique rigoureuse.
Ainsi, l’équipe britannique a entrepris d’élargir leur panel de patients présentant des mutations sur ce gène, et a par ailleurs sollicité une collaboration avec l’équipe de Cyril Bourgeois du Laboratoire de Biologie et Modélisation de la Cellule de l’ENS, qui travaillait déjà sur le gène DDX17. L’objectif de cette collaboration était d’établir un lien entre la fonction de ce gène dans le développement neuronal et cette nouvelle pathologie, afin de mieux en comprendre les causes au niveau cellulaire et moléculaire.
Une signature transcriptomique associée au neurodéveloppement
L’équipe de Cyril Bourgeois a tout d’abord mené une analyse transcriptomique sur des cellules humaines. Cette approche consiste à séquencer de manière exhaustive tous les ARN messagers de ces cellules, en comparant celles où le gène DDX17 est inactivé à celles où il ne l’est pas. Cette analyse, réalisée par une doctorante de l’équipe, Valentine Clerc, et par un ingénieur bio-informaticien, Xavier Grand, a permis d’identifier 350 gènes dont l’expression est dérégulée en l’absence du gène DDX17.
Une grande majorité de ces gènes est associée au développement morphogénique, et plus spécifiquement au développement neuronal, fournissant une piste sur le mécanisme pathologique sous-jacent chez les patients.

Des modèles animaux pour comprendre la fonction du gène et les effets des mutations
Fort de ces premières constatations, des recherches expérimentales sur des modèles animaux ont été réalisées afin de mieux comprendre les fonctions de DDX17 dans le développement cérébral.
Tout d’abord, Julien Courchet et son équipe ont utilisé une technique de micro-injection pour introduire, dans le cerveau d’embryons de souris en gestation, des molécules visant à inactiver ponctuellement le gène DDX17, sans le supprimer entièrement du génome, contrairement à un knock-out. Cette approche, plus rapide, permet d’obtenir des résultats en quelques semaines, contre plusieurs mois pour un knock-out. En suivant l’évolution du cerveau des souris de la période prénatale à la phase postnatale, Géraldine Meyer-Dilhet, ingénieure d’étude au sein de l’équipe, a observé des anomalies dans la croissance et la ramification des axones, signes d’un développement neuronal altéré.
Ces observations ont été confirmées par des expériences in vitro : Géraldine Meyer-Dilhet et Laloe Monteiro, deux ingénieures de l’équipe, et Martijn Kerkhofs, chercheur post-doctorant, ont mis en place des cultures neuronales issues de souris, dans lesquelles ils ont reproduit l’inhibition du gène en question. Les mêmes anomalies de croissance et de ramification ont été constatées, confirmant que l’altération se manifeste à la fois in vivo et in vitro.
Pour approfondir cette étude, des expériences de « rescue » (ou de sauvetage) ont aussi été réalisées: une copie fonctionnelle du gène DDX17 a été ré-introduite dans les cellules après son inactivation, ce qui a restauré le développement neuronal typique. Ces résultats renforcent l’idée que les altérations observées sont directement liées à l’inactivation du gène DDX17.
De manière complémentaire, une équipe de l’université de Portsmouth a réalisé un knock-out du gène DDX17 chez le crapaud xénope, un modèle animal particulièrement adapté à ce type de manipulation. Cette inactivation a révélé des anomalies similaires à celles observées chez la souris, notamment une croissance neuronale et axonale réduite.
De plus, une analyse comportementale des têtards dépourvus du gène a pu être réalisée. Grâce à des tests en labyrinthes, l’équipe anglaise a mis en évidence des altérations : alors que les têtards normaux alternent systématiquement entre les directions gauche et droite, ceux privés du gène DDX17 montrent un comportement désorganisé. Ces résultats traduisent une altération significative de leur mémoire de travail.
Bien qu’une comparaison directe avec les humains reste limitée, ces résultats renforcent l’hypothèse d’une implication du gène DDX17 dans les troubles du neurodéveloppement, accompagnée de répercussions comportementales.
En effet, le gène DDX17 est hautement conservé parmi les eucaryotes, c’est-à-dire qu’il est présent chez tous les organismes multicellulaires, depuis la levure jusqu’à l’humain. Cette conservation exceptionnelle tout au long de l’évolution met en évidence son importance biologique fondamentale.
Vers de nouvelles perspectives diagnostiques et thérapeutiques
Cette étude ouvre la voie à de nombreuses applications. « Notre objectif est d’inciter les cliniciens à intégrer le gène DDX17 dans leurs analyses génétiques afin d’identifier d’autres porteurs de mutations et de mieux comprendre les conséquences cliniques associées », souligne Cyril Bourgeois. À ce titre, une équipe néerlandaise gérant une base de données dédiée aux maladies génétiques rares (Human Disease Genes) a récemment pris contact avec lui afin de lui proposer d’être la personne référente pour le suivi et l’analyse des futurs cas cliniques identifiés. Cette plateforme permet de mettre en relation les familles de nouveaux patients avec des professionnels de santé et des chercheurs. D’un point de vue fondamental, les recherches futures viseront à décrypter les mécanismes par lesquels ces mutations altèrent la fonction de la protéine. De plus, plusieurs revues internationales ont mis en évidence le lien entre la famille de protéines à laquelle appartient DDX17 et les troubles du neurodéveloppement, ouvrant ainsi un champ d’investigation prometteur.
Une collaboration internationale exemplaire
Ce projet a impliqué quatre unités de recherche Inserm (Laboratoire de Biologie et Modélisation de la Cellule de Lyon, le laboratoire de Physiopathologie et Génétique du Neurone et du Muscle de Lyon, le Centre de Recherche en Cancérologie de Lyon, le laboratoire de Génétique des Anomalies du Développement de Dijon) et de nombreux partenaires à travers le monde. Il illustre la puissance de la science collaborative pour répondre à des questions complexes. La publication de ces travaux dans une revue prestigieuse souligne leur importance et leur potentiel impact pour les patients et les cliniciens.
Cette étude n’est qu’un premier pas dans l’exploration des rôles du gène DDX17 et ouvre la voie à des avancées significatives dans le diagnostic et la prise en charge des troubles du neurodéveloppement.
En résumé
En savoir plus:
Bonne nouvelle

A lire également
De la recherche fondamentale au patient, il n’y a parfois qu’un pas.
À l’occasion de la Journée Internationale des Maladies Rares, Thomas Boulin nous explique le lien entre troubles du neurodéveloppement, épilepsies sévères et le gène NBEA.
En savoir plus
IMINDgénétique
Sommaire
De la recherche fondamentale au patient, il n’y a parfois qu’un pas.
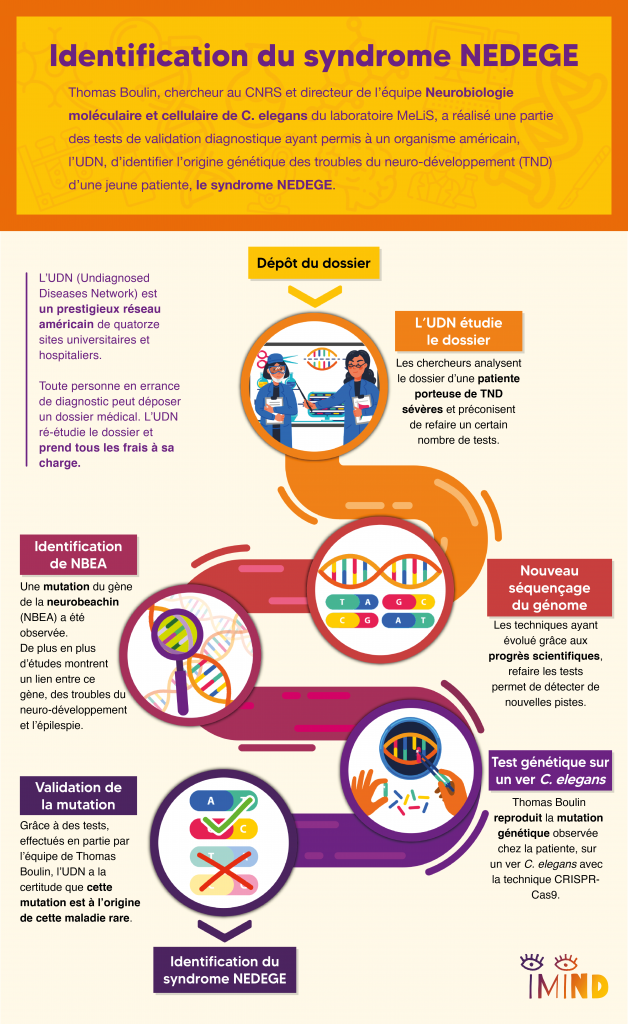
Thomas Boulin, chercheur au CNRS et directeur de l’équipe « Neurobiologie moléculaire et cellulaire de C. elegans » du laboratoire MeLiS, a réalisé une partie des tests de validation diagnostique qui a permis à un organisme américain, l’UDN, d’identifier l’origine génétique des troubles du neuro-développement (TND) d’une jeune patiente, le syndrome NEDEGE qui résulte d’une mutation du gène NBEA. Lui qui d’habitude se passionne pour des questions de recherche fondamentale, a éprouvé un regain d’enthousiasme à pouvoir appliquer sa recherche pour confirmer le diagnostic d’une jeune patiente américaine.

« La recherche, ce n’est pas un chercheur qui se lève le matin en décidant d’étudier une pathologie pour savoir comment elle fonctionne. Ça ne se passe pas comme ça, sinon, on n’aurait plus de cancer ».
C’est avec ces mots que Thomas Boulin a commencé son récit. En effet, la recherche scientifique reste un mystère pour beaucoup. On distingue la recherche fondamentale, qui vise à comprendre les phénomènes biologiques et dont le but est le progrès de la connaissance, à laquelle on oppose souvent, de façon erronée, la recherche appliquée dont le but est de répondre à une question précise, par exemple clinique. Alors que le financement de la recherche se fait aujourd’hui essentiellement par appels d’offres ciblés, la recherche fondamentale est davantage critiquée sur son utilité, en comparaison à la recherche appliquée, plus concrète à première vue. Or, sans recherche fondamentale, pas de recherche appliquée car celle-ci s’appuie sur le socle de connaissance issu de la recherche fondamentale. L’équipe de Thomas Boulin du laboratoire MeLiS étudie le fonctionnement des canaux potassiques qui régulent l’activité électriques des neurones. En d’autres termes, il cherche à comprendre les conditions nécessaires, au niveau moléculaire, pour que l’information circule correctement dans nos réseaux neuronaux. Ses travaux se situent donc dans le champ de la recherche fondamentale. C’est pourtant grâce à ces travaux qu’il a pu développer un nouvel outil diagnostic pour le syndrome NEDEGE.
Le parcours diagnostic dans les maladies rares
Beaucoup de maladies rares ont une origine génétique. Le parcours patient classique consiste à faire des examens génétiques pour identifier des gènes-candidats, c’est-à-dire des gènes comportant une mutation qui pourrait être à l’origine de la maladie rare. En plus du génome du patient, on séquence donc le génome des parents car ces mutations apparaissent fréquemment après la fertilisation de l’ovocyte par un accident génétique lors de la reproduction de l’ADN. On dit qu’il s’agit de « mutations de novo ». Aucun des deux parents n’ayant la mutation, la comparaison du génome des parents et de l’enfant permet d’identifier cette liste de gènes-candidats. Souvent, le parcours médical s’arrête là car les médecins n’ont pas forcément les savoirs, ni les outils technologiques pour aller plus loin. C’est là qu’entre en scène Hugo Bellen, un généticien de la mouche Drosophile et son réseau américain, UDN, Undiagnosed Diseases Network, dont l’originalité est de mettre des plateformes technologiques de pointe au service du diagnostic génétique et de démontrer qu’une mutation est bien à l’origine de la maladie du patient.
Qu’est-ce que l’UDN?
 L’UDN, pour Undiagnosed Diseases Network, est un réseau américain de quatorze sites universitaires et hospitaliers les plus prestigieux du pays, financé par le NIH. Un patient, ou sa famille, en errance diagnostique peut déposer auprès de cet organisme un dossier composé de tous ses antécédents et tests médicaux. Pour les patients pris en charge, l’UDN étudie tous les éléments du dossier et va actualiser certains tests ou faire des tests complémentaires, à la lumière des avancées technologiques et scientifiques les plus récentes. C’est entièrement pris en charge par le réseau, ce qui n’est pas négligeable lorsqu’on connaît le coût des frais médicaux aux États-Unis.
L’UDN, pour Undiagnosed Diseases Network, est un réseau américain de quatorze sites universitaires et hospitaliers les plus prestigieux du pays, financé par le NIH. Un patient, ou sa famille, en errance diagnostique peut déposer auprès de cet organisme un dossier composé de tous ses antécédents et tests médicaux. Pour les patients pris en charge, l’UDN étudie tous les éléments du dossier et va actualiser certains tests ou faire des tests complémentaires, à la lumière des avancées technologiques et scientifiques les plus récentes. C’est entièrement pris en charge par le réseau, ce qui n’est pas négligeable lorsqu’on connaît le coût des frais médicaux aux États-Unis.
Dossier UDN N°068
Bien que doté de moyens très importants, ce réseau s’appuie aussi sur l’expertise de collaborateurs internationaux et lance régulièrement des appels à la communauté scientifique. C’est ainsi qu’un jour apparaît sur ce portail le cas d’une jeune fille américaine porteuse d’un trouble du neuro-développement sévère avec un trouble du développement intellectuel et des crises d’épilepsie fréquentes pour laquelle l’UDN a identifié le gène Neurobeachin (NBEA) comme gène-candidat principal. En effet, une étude de 2019 très récente avait montré un lien entre les troubles du neuro-développement, l’épilepsie et ce gène. Quand Thomas voit cet appel, ça fait tilt !
Il se trouve que Sonia El Mouridi, doctorante dans l’équipe, avait découvert un rôle nouveau de la Neurobeachin dans les processus biologiques qui intéressent l’équipe. N’étant pas le cœur de son projet de doctorat, cette observation était simplement présentée dans une annexe de sa thèse sans avoir été formellement publiée. Qui aurait pu prédire que, quelques années plus tard, forte de ces résultats et de son expertise sur le ver C. elegans, l’équipe de Thomas allait pouvoir réaliser pour l’UDN une partie des tests de validation fonctionnelle démontrant que cette mutation inconnue jusque-là était bien à l’origine des troubles de la patiente et aider ainsi à confirmer le diagnostic génétique de la maladie rare de cette jeune fille.
Comment l’équipe s’y est-elle prise ?
Un modèle animal sur mesure
Pour démontrer l’effet délétère d’une mutation identifiée chez un patient, il est très souvent nécessaire de passer par des modèles animaux. La souris serait tout à fait pertinente mais cela reste extrêmement cher et long et surtout, impossible à réaliser à grande échelle. L’approche préconisée par l’UDN et Thomas Boulin s’oriente donc vers des modèles animaux plus simples, moins coûteux et où le temps de génération est plus rapide. Ainsi la drosophile, le poisson zèbre, mais aussi le ver C. elegans sont des modèles de choix. On est en droit de se demander en quoi la drosophile ou le ver C. elegans sont des modèles pertinents pour étudier l’humain étant donné que nous n’avons pas grand-chose en commun à première vue. Il s’avère en réalité, que notre patrimoine génétique n’est pas si différent. Les gènes affectés dans les maladies rares sont souvent des gènes très importants, maintenus au cours de l’évolution, et que l’on retrouve chez l’humain, mais aussi chez la drosophile ou le ver C. elegans. C’est ce qu’on appelle la conservation évolutive.
Qu’est-ce que C. elegans ?
Le nématode C. elegans est un ver d’un millimètre de long, à peine visible à l’œil nu. Il est l’un des modèles animaux utilisés en biologie depuis 50 ans pour disséquer les processus biologiques fondamentaux, comme par exemple la mort cellulaire programmée qui est impliquée dans le cancer. Cet « organisme modèle pionnier » est très prisé pour les études génétiques du fait de sa robustesse, de son temps de génération extrêmement rapide (3 jours), des nombreux outils génétiques disponibles et de son coût très limité.
La révolution CRISPR-Cas9
Récapitulons : nous avons une patiente, un gène candidat, NBEA, et un modèle animal, le ver C. elegans, spécialité de l’équipe de Thomas. La première étape a donc consisté à reproduire la mutation identifiée chez la patiente dans le génome de C. elegans afin de créer un « modèle sur mesure ». Grâce à la technologie des « ciseaux moléculaires » CRISPR-Cas9, on peut aujourd’hui d’introduire une cassure dans un gène cible afin de forcer la cellule à la réparer avec une séquence génétique artificielle, contenant la mutation. Avant de mettre au point cette technologie, on avait tendance à surexprimer les protéines mutées, ce qui a souvent des effets délétères et peut entraîner des artefacts. L’approche CRISPR-Cas9 permet maintenant de faire des modifications génétiques de façon très fine, sans modifier ce qu’il y a autour. On peut ainsi étudier l’impact d’une mutation dans un modèle animal de la façon la plus fidèle possible.
La validation de mutation
La Neurobeachin est une protéine immense composée de 2500 lettres, ou acides aminés. Or, la mutation de la patiente n’affecte qu’un seul de ces acides aminés, et ce, en opérant un simple remplacement par un autre acide aminé. Or les dysfonctionnements produits par ce type de substitutions sont presque impossibles à prédire théoriquement. Grâce aux animaux génétiquement modifiés, Thomas et son équipe ont donc procédé à différents tests fonctionnels pour vérifier l’impact de la mutation sur le fonctionnement de la Neurobeachin. Grâce à ces tests, l’UDN a été en mesure de conclure avec certitude que cette mutation sur le gène NBEA de la patiente était bien à l’origine de sa maladie rare.
De l’importance d’identifier ces syndromes
Valider l’impact d’une mutation et mettre un nom sur un syndrome permet d’apporter la certitude au patient, à ses proches et à l’équipe médicale que la pathologie est bien liée à la mutation d’un gène. L’errance diagnostique prend fin, ce qui représente souvent un grand soulagement psychologique pour la famille. Une fois que le diagnostic a été posé, les personnes concernées par ce syndrome peuvent se rencontrer : d’une part pour échanger sur leur quotidien, se conseiller, s’épauler et d’autre part pour avoir une idée de l’évolution de la maladie. Ils peuvent aussi se regrouper en associations dans le but de communiquer et de lever des fonds pour encourager les travaux de recherche dans ce domaine. Il y a sans doute d’autres patients dans le monde avec une mutation de la Neurobeachin, dont on pense simplement qu’ils sont épileptiques ou qu’ils ont un trouble du neuro-développement. Le véritable enjeu aujourd’hui est d’associer les deux, troubles du neuro-développement et Neurobeachin, pour que les généticiens testent aussi ce nouveau gène si les symptômes sont concordants.
Que sait-on sur la Neurobeachin (NBEA)?
Il existe encore peu d’informations sur cette protéine. Elle est essentiellement exprimée dans le cerveau et joue vraisemblablement un rôle très important dans les systèmes de contrôle de l’activité cérébrale. NBEA a été identifié comme gène candidat pour des maladies avec TND en 2003. Une simple mutation de ce gène peut entraîner des conséquences très sévères du point de vue neuro-développemental. Certains gènes supportent facilement des mutations dans leur séquence. D’ailleurs, si l’on compare le génome de deux personnes lambda, on va s’apercevoir qu’il y a beaucoup de différences entre elles, ce qui est tout à fait normal. Mais certains de nos gènes supportent moins bien les mutations. NBEA est un exemple de ces gènes extrêmement contraints qui ne tolèrent presque aucune mutation.
Depuis, l’équipe de Thomas a décroché un financement sur 4 ans de l’Agence Nationale de Recherche pour mieux comprendre les bases moléculaires et cellulaires du fonctionnement de la NBEA. Il collabore aussi avec Tristan Sands de l’Université de Columbia sur la validation diagnostique chez de nouveaux patients.
« Le travail de Sonia n’avait pas du tout pour finalité de résoudre l'errance diagnostique de cette petite fille. Or c’est ce qui s’est passé, l’histoire est belle et donne du sens à notre recherche. Et cela montre à quel point la recherche fondamentale est nécessaire et importante, et qu’il ne faut pas être trop contraint dans nos idées. », souligne Thomas.
En résumé